
Cellular responses to DNA damage: mechanistic insights and clinical applications
 is disabled.")
The IGMM lecture theatre was packed with people on the 12th November, eager to listen to Steve Jackson’s seminar, which was entitled ‘Cellular responses to DNA damage: mechanistic insights and clinical applications’. I did my undergraduate project in Steve’s lab two years ago, so I was interested to see what they’ve been up to since I left and started my PhD here at the IGMM. Steve has had a very exciting career – as well as leading his own lab at the Gurdon Institute in Cambridge for 28 years he has also started several biotech companies. One of these, KuDOS Pharmaceuticals, was sold to AstraZeneca in 2006 for £120 million.
Steve has built his career on investigating the cellular response to DNA damage. Every day the DNA in each of our cells experiences tens of thousands of lesions1. Thankfully, the cell has multiple pathways by which different types of DNA damage are repaired. Steve’s lab primarily focuses on double-strand breaks, which can be repaired by either the homologous recombination (HR) or non-homologous end joining (NHEJ) pathways. If DNA damage is not repaired then this can lead to devastating consequences for the cell – defects in the response to DNA damage are the cause of many diseases, such as Ataxia-Telangectasia and Bloom’s syndrome, both of which make patients more likely to develop cancer. For this reason, it seems counter-intuitive that inhibiting DNA damage repair would be a strategy for treating cancer. However, Steve’s first company, KuDOS, developed olaparib, a drug which inhibits PARP, a protein involved in repairing single-strand breaks in the DNA.
Olaparib’s success rests on the principle of ‘synthetic lethality’. PARP inhibition results in cell death in cells that are mutated in BRCA1 or BRCA2, proteins involved in repair of double-strand breaks via the HR pathway. Normal cells however are not killed by PARP inhibition. Olaparib is a marketed drug for ovarian cancer in multiple countries, and is in late stage clinical trials for the treatment of several more cancers, including breast, gastric and prostate. Steve also suggested that it could one day be used prophylactically in BRCA-mutant patients to kill off BRCA mutated cells before they can develop into tumours.
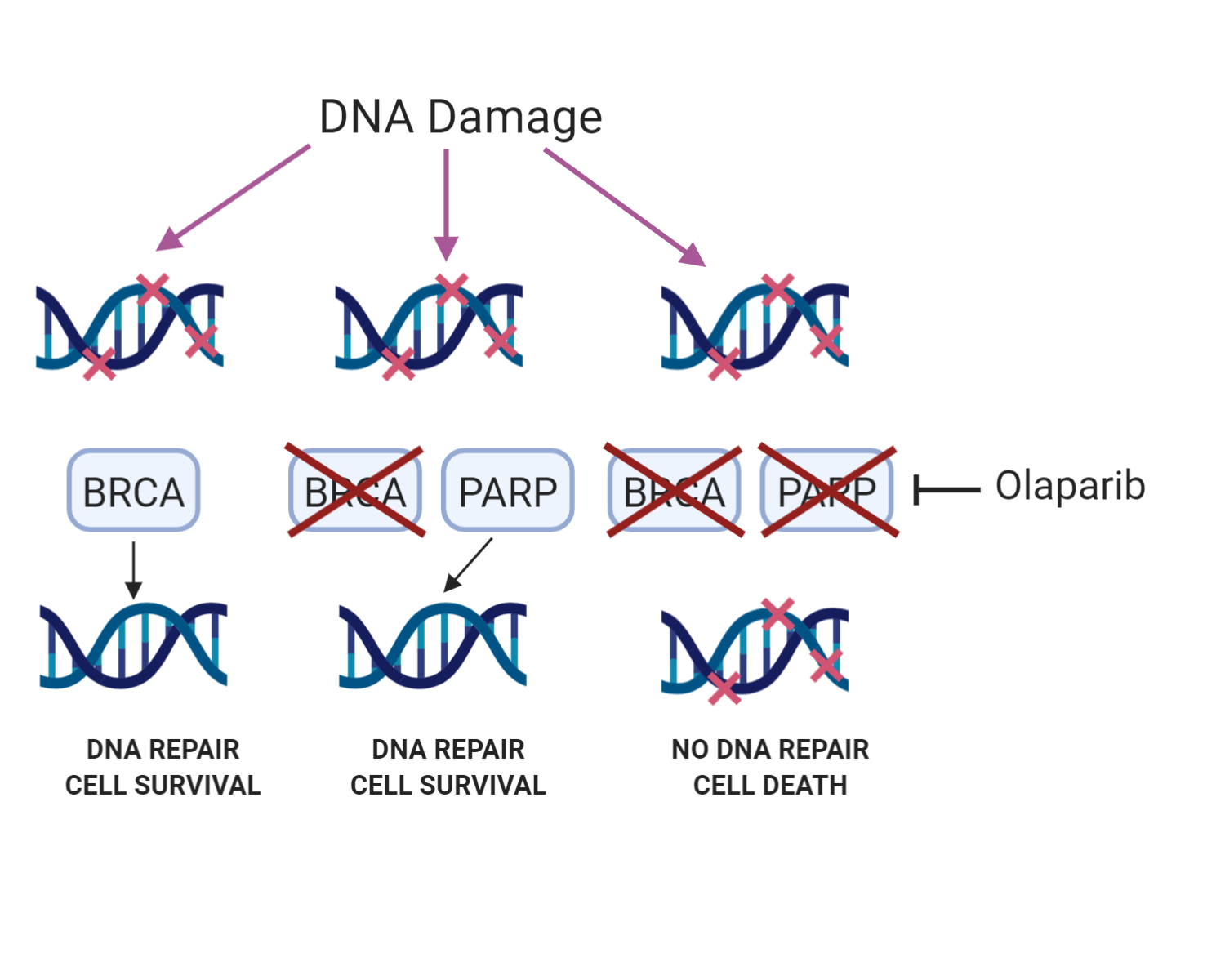
The principle behind using PARP inhibitors to treat cancer in BRCA-mutant cells. Blocking DNA repair with a PARP inhibitor kills the cell when the back-up pathway (BRCA) is disabled. Made with BioRender.
However, not all patients respond to olaparib and like with all targeted cancer therapies, many become resistant. Steve’s lab are now investigating resistance mechanisms in an effort to better treat cancer patients, primarily by using large-scale CRISPR screens. For example, a screen they carried out in BRCA1-deficient breast cancer cells treated with PARP inhibitors identified a new complex made up of two proteins, FAM35A and C20orf196, which they (and several other groups which discovered the complex at the same time) called Shieldin2. Shieldin inactivation causes strong PARP inhibitor resistance. However, they also found that Shieldin inactivation confers sensitivity to cisplatin, suggesting future treatment strategies if PARP-inhibitor-treated patients present with Shieldin inactivating mutations in the clinic.
Cancers deficient in another protein involved in double-strand break repair, ATM, are also being treated with PARP inhibitors, which raises the question of whether the resistance mechanisms are similar. To investigate this, the lab carried out another screen in mouse embryonic stem cells deficient in ATM, which they treated with the DNA topoisomerase I inhibitor topotecan3. They found that knocking out several different proteins involved in NHEJ repair or alternatively knocking out the BRCA1-A complex could overcome the increased sensitivity to topotecan caused by ATM knockout. A similar effect was seen when the cells were treated with PARP inhibitors. This suggests that suppressor mutations in an ATM-mutant background are different to those in a BRCA1-mutant background, which could present a new opportunity for patient stratification.
Steve then went on to talk about some work on the MDC1 protein4, which was primarily carried out by Israel Salguero, my day-to-day supervisor when I was in the lab. MDC1 is known to act as a ‘mediator’ of the DNA damage response. At double-strand break sites histone H2AX is phosphorylated, and MDC1 binds to this phosphorylation and recruits further proteins involved in the DNA damage response. However, H2AX and MDC1 deficient mice are not equivalent phenotypically – H2AX deficient mice are only tumour prone in a p53 null background, while MDC1 knockout mice are tumour prone even in the presence of p53 function. This suggests MDC1 may have an H2AX independent role, which is what Steve’s lab set out to investigate. They found that MDC1 knockout human cells are more sensitive to ionising radiation than H2AX knockout cells, further suggesting MDC1 function does not wholly depend on H2AX. In addition, they found that MDC1 recruits 53BP1, another DNA repair factor, to double-strand break sites in an H2AX-independent manner. This requires the PST region of MDC1, a region of the protein whose role in the DNA damage response had until then not been known. This PST-repeat region also binds to nucleosomes which is vital for localising the DNA damage response to chromatin in the absence of H2AX.
Overall, the talk was an enjoyable mix of basic DNA repair biology and its applications to drug discovery and development, which is what originally attracted me to Steve’s lab. Steve has recently started another biotech company, Adrestia Therapeutics5, which aims to restore the biological balance in dying cells through applying the principle of synthetic viability – it will be exciting to follow what happens to this company, and what Steve gets up to next.
References
1. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009) doi:10.1038/nature08467
2. Dev H, Chiang TWW, Lescale C, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 20, 954–965 (2018) doi:10.1038/s41556-018-0140-1
3. Balmus G, Pilger D, Coates J, et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat Commun. 10, 87 (2019) doi:10.1038/s41467-018-07729-2
4. Salguero I, Belotserkovskaya R, Coates J, et al. MDC1 PST-repeat region promotes histone H2AX-independent chromatin association and DNA damage tolerance. Nat Commun. 10, 5191 (2019) doi:10.1038/s41467-019-12929-5
5. ADRESTIA THERAPEUTICS. https://www.adrestia.com/ Accessed November 22, 2019.
(Bethany Bartlett, made with BioRender)

